Before You Begin: Design and Genotyping
- Design: We strongly recommend that we are involved in the design of the targeting strategy. The efficiency and thus the likelihood of success depends critically on the design. Mistakes in mice are expensive in terms of both money and time.
- Genotyping: Design of a genotyping strategy is an integral part of the design process--another reason to involve us in the design process. The mice we give you will be useless if you can't identify which carry the desired mutation.
- No passenger mutations: An additional consideration is that some of the repair pathways engaged by CRISPR/Cas are error-prone, in particular those engaged when single-stranded oligos are used to incorporate missense mutations. In these cases, we recommend careful characterization of the targeted gene in the progeny of founder mice mated to wild type, to ensure that there are no second-site mutations in the same gene at a distance from the intended change.
CRISPR/Cas Reagents For Injection
- Reagents for injection are commercially prepared. Contact us for details.
- The current commercial vendor costs for injection-ready reagents (guide RNA, Cas9 protein and ssDNA oligo or dsDNA targeting vector) are from $1000 to $2000.
CRISPR/Cas Mutagenesis by Injection of Fertilized Eggs
What We Will Do Part I- We will advise you. Early in your planning, please consult with Ron Conlon and/or David LePage to discuss your project.
- An account number.
- We require a demonstration that the guide RNA(s) directs sequence-specific cutting in an in vitro assay.
- We will inject fertilized mouse eggs with the reagents.
- While we don't provide a guarantee, our goal is to deliver enough mice to generate the desired mutant mice.
- We will house the mice until they are safe to transport.
- You will receive and house the mice.
- You will genotype the mice we provide, and tell us which mice and how many are mutant.
- We ask that you acknowledge the contribution of the Case Transgenic and Targeting Facility in seminars and publications.
- Investigators need an IACUC protocol from their institution to receive mice from the core. CWRU IACUC protocols need to specify use of the Case Transgenic Core. Investigators normally do not need an IBC protocol of their own for this work: the review for recombinant DNA in animals will be conducted on the information submitted through online ordering for the transgenic core, and will be added to the core's IBC protocol as an amendment if approved. You may be contacted by the IBC for additional information as part of rDNA review after submission of the order. IBC approval is required prior to initiation of injections.
- Currently (7/28/26), the core is injecting immediately after reagents are available and the project is ready. During this time mice to fulfill the service are ordered and received from Jax.
- From the injection date, it will be a minimum of a further 6 weeks before we will be able to deliver mice to you (almost 3 weeks for gestation, and at least 3 weeks of postnatal growth before weaning).
- $5,500 and the cost of donor females for CRISPR/Cas9 on the B6SJL background. The donor female cost ranges from $1,000-$2,000.
- $7,000 and the cost of donor females for CRISPR/Cas9 on C57Bl/6J inbred background. The donor female cost ranges from $1,000-$2,000.
- Local, non-CWRU orders will be charged an additional $300 for packing and transporting mice. Non-local orders will be charged $300 and the costs of transportation.
CRISPR Mutant Mouse Ordering
- PI Name, Address and Contact Info
- Account Number
- IACUC Protocol Number
- Project name
- Mutation Type
- Mouse Strain to Inject
- Is a Phenotype expected?
- Building and Room to Deliver Mice to
- Purpose and design of the animal experiments
- Lay description of the animal experiments
- Name and function of the gene
- Whether the gene is involved in infectious disease in normal healthy adult humans
- Whether the gene presents any hazard to health or the environment
- A .pdf file diagram the targeting strategy
CRISPR/Cas Success Rates
We have completed nearly 200 different CRISPR knockin/out projects. The vast majority have generated multiple independent founders with the desired allele. Typically founder mice are not mosaic for the founder allele, unlike in early applications of this technology. We have successfully generated many knockout, point mutation knockins, reporter/tag/Cre knockins, floxed or conditional alleles, Rosa26 and H11 safe harbor targeted transgenes, and floxed-stop alleles.Mutation and genotyping strategies
- Point mutations will be made with a single guide RNA, the nuclease version of Cas9 and a single-stranded oligonucleotide. For success, it is important that 1) the gRNA cuts close (10bp or less) to the desired mutation 2) the gRNA has been demonstrated to be active and 3) the recombinant stop continued cutting through mutation of the PAM or gRNA seed. Genotyping of founders should be performed by deep sequencing (MiSeq) of about 200 bp amplified around the mutation. Progeny of the founders (F1s) can be genotyped by PCR, but because the repair mechanisms engaged are error-prone, the locus should be sequenced in F1 mice to ensure that there are no nearby passenger mutations in the mutated gene.
- Knockouts (null alleles) will be made with two gRNAs and Cas9 nuclease to delete an essential exon or exons, ideally the functional core of the protein, in a manner predicted to result in out-of-frame splice products after deletion. Deletion of exon(s) can be detected in founders by multiple different pairs of PCR primers. Transmission of the same allele should be confirmed with multiple sets of primers in the progeny of the founders. Subsequent genotyping can be by a simple PCR assay specific to the mutated allele.
- Insertions will be made with a single guide RNA, Cas9 nuclease, and a double-stranded DNA vector with 1kb arms of homology. Both random and targeted insertions are possible in founders. The combined random and targeted insertions first should be detected by PCR with primers internal to the construct. Targeted insertions then can be identified by PCR with one primer in the insertion, and the other primer in the endogenous locus outside the arm of homology. Assays for targeting need to be performed on both sides of the insertion to confirm proper targeting. It is possible for founders to carry both targeted and random insertions. These events can only be detected in the progeny of the founders, by again genotyping for both internal primer pairs, and pairs which can detect gene targeting. Once progeny transmit only targeted alleles, a simpler genotyping strategy with only internal primers can be used.
- Replacements, including floxed alleles, will be made with 2 guide RNAs, Cas9 nuclease, and a double-stranded DNA vector with 1kb arms of homology. First, random and targeted insertions in founders can be detected by PCR with primers internal to the construct. Then targeted insertion can be identified in founders by PCR with one primer in the insertion, and one primer in the endogenous locus outside the arnm of homology, on both sides of the insertion. As for insertions above, random and targeted integrants both can be present in a founder, so genotyping to distinguish random from targeting needs to continue until a targeted mouse stops transmitting random integrants.
- If you are experiencing problems with genotyping founders, please contact us. It is better to consult sooner rather than later, before your founders age out. We are here to help, and want your project to succeed.
Workflow
- You will provide us with annotated mouse sequences for the genomic locus and the mutation you want generated. We will identify candidate guide RNAs for your mutation. If potential off-target sites are not on the same chromosome as your gene, any off-target mutations will segregate away through meiosis when you breed the mice. All the off-target sites should not be in genes.
- We will ask you to identify which candidate gRNAs are active with an in vitro cutting assay. gRNAs generated with the kit are assayed by incubation with Cas9 protein provided by the kit and your amplified genomic target, then running the products on an agarose gel like a restriction nuclease assay. Run uncut target genomic fragment in one lane and the different gRNA assays on the same gel to determine which gRNAs most efficiently direct cutting--the majority are active, you want to avoid the ones which don't work. In our experience, this in vitro cleavage assay correlates better with gene targeting in injected eggs than the predictions of online tools or cell transfection assays.
- Once active guide RNAs have been identified, we will develop a detailed targeting strategy.
- For point mutations, once an active gRNA which cuts close the desired mutation (ideally 10bp or less) has been identified, design silent passenger mutations that disrupt the PAM (preferred) or gRNA seed (the 10 nucleotides closest to the PAM) to prevent recutting of recombinants. If your substitution generates a mutation in the PAM or seed sequence, it may not be necessary to incorporate passenger mutations. When designing synonymous passenger mutations, be certain to avoid rarely used codons using a mammalian codon bias table. It is possible to recover desired edits without destroying the PAM or seed sequence, but the yield can be much lower because of recutting and mutagenesis of already recombined alleles. Single-stranded oligos of 100bp centered on the mutation are sufficiently long for point mutations.
- Purchase the reagents. We purchase our reagents (guide RNAs and Cas9 protein and PAGE-purified oligo) for injection of fertilized eggs from a commercial vendor.
- You will genotype the founder mice. Developing PCR genotyping assays for founder mice is difficult due to the vagaries of PCR, the absence of natural positive controls, and the occasional low representation of the mutation in the mice. These three issues interact. The best way to optimize PCR is to try multiple different primer pairs. In order to know if a primer pair works, you need to generated a robust positive control. Because of mutation mosaicism in founders that postive control should be able to detect 0.1 copies (or below) of the mutation per diploid genome in the context of wild type genomic DNA.
- It may take several tries to find primer pairs which work well in the context of mouse genomic DNA--empirical testing of different primer pairs is the single most important step in optimizing a genotyping assay. To validate a PCR assay, a negative control of wild type mouse genomic DNA and positive controls of wild type mouse genomic DNA spiked with targeting vector at 1, 0.1, 0.01, and 0.001 copies per diploid genome should be performed. This series will demonstrate the sensitivity of the assay and reveal background problems--a point of no detection should be reached. It is not adequate to test the PCR assay on dilutions of construct DNA in the absence of genomic DNA. Markers should be run on the same gel in order to determine if the amplified product is of the correct size. The number of cycles should be no more than 30-40, and should be performed on no more than 25-50ng of genomic DNA. If the PCR reaction shows a smear in the lane of the gel, lower the amount of input DNA.
- A calculator for vector mass to obtain one copy per diploid genome is here.
- Treat each founder mouse as the start of an individual mutant line. You should characterize (at least preliminarily) multiple independent mutations.
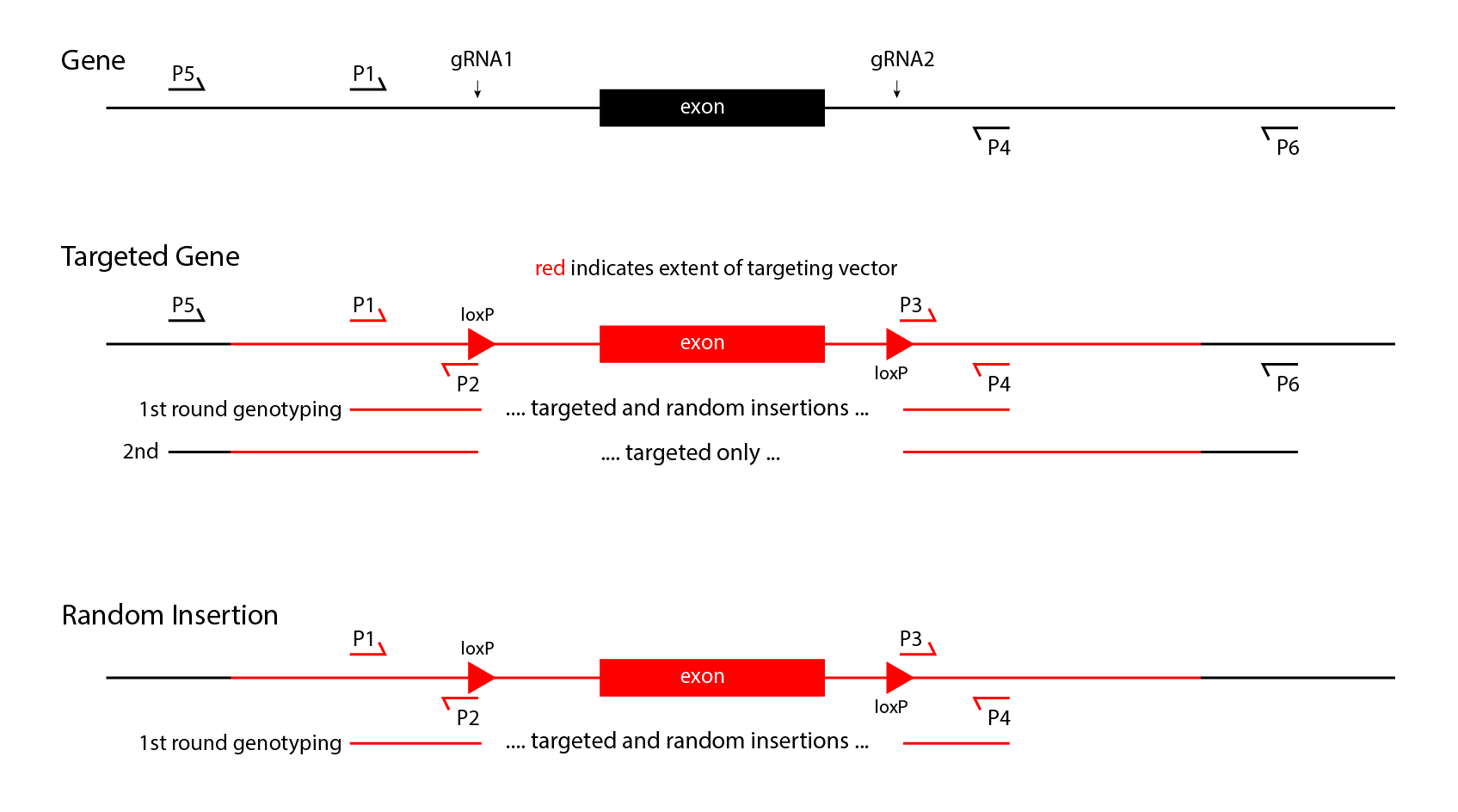
A 2-stage strategy to detect floxed alleles in founder mice is diagrammed above. Note that this process requires finding 4 different sets of primer pairs that can detect 0.1 copies per diploid genome. The positive controls for the short product PCR assays internal to the construct can use dilutions of the targeting vector spiked into wild type genomic DNA. DNA to serve as positive controls can be obtained as synthetic, uncloned, double-stranded DNA from IDT, GeneWiz, GeneArt or others for a reasonable price (about $200 per construct).
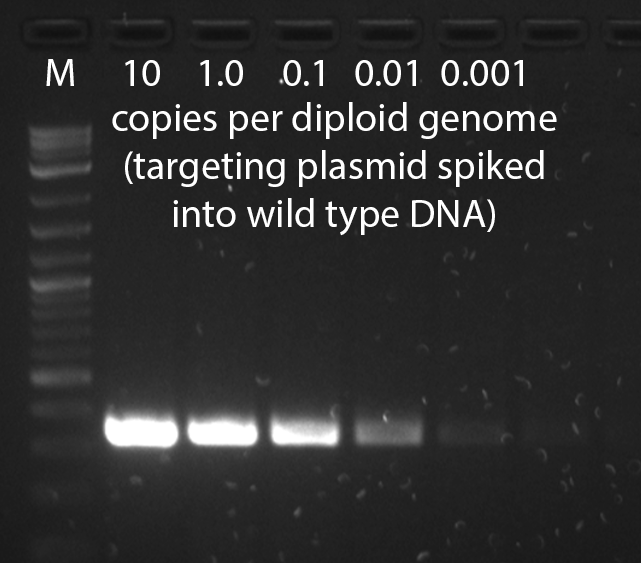
A gel from a positive control dilution series PCR genotyping assay that has high sensitivity, able to detect less than one copy per diploid genome.